Diagnosing Sickle Cell Anemia
Diagnosis is a critical step in managing sickle cell anemia. Here’s how medical professionals determine the presence of this condition and assess related risks.
Blood Testing for Hemoglobin Variants
- Newborn Screening: In the U.S., newborns are routinely tested for sickle cell anemia.
- Older Children and Adults: The test is also available for older individuals, where blood is drawn from a vein or, in the case of young children and babies, from a finger or heel.
Laboratory Analysis
The collected blood sample is analyzed in a lab to detect the presence of sickle cell hemoglobin.
Additional Testing for Complications
- Further Examinations: If sickle cell anemia is confirmed, healthcare providers may recommend tests to identify potential complications.
- Genetic Counseling: Carriers of the sickle cell gene are often advised to consult a genetic counselor.
Stroke Risk Assessment in Children
- Ultrasound Testing: A non-invasive ultrasound can measure cerebral blood flow to gauge stroke risk.
- Age of Testing: Suitable for children starting at age 2.
- Preventive Measures: Regular blood transfusions can mitigate the risk of stroke.
Prenatal Diagnosis
- Amniotic Fluid Sampling: Sickle cell disease can be diagnosed prenatally by testing the amniotic fluid.
- Parental Testing: Parents with sickle cell anemia or the trait should discuss prenatal screening options with their healthcare provider.
Treatment and Management of Sickle Cell Anemia
Effective management of sickle cell anemia focuses on minimizing pain, alleviating symptoms, and thwarting complications. Here’s a breakdown of the current treatment modalities:
Medications
- Hydroxyurea (Droxia, Hydrea): Administered daily, hydroxyurea can lessen pain crises and potentially reduce the need for blood transfusions and hospitalizations. However, it carries a heightened risk of infections and is not recommended during pregnancy.
- L-glutamine Oral Powder (Endari): This medication aids in decreasing the frequency of pain crises.
- Crizanlizumab (Adakveo): Suitable for adults and children over 16, this injectable drug can help lower the number of pain crises. Possible side effects include nausea, joint pain, back pain, and fever.
- Voxelotor (Oxbryta): Prescribed for adults and children over 12, voxelotor is taken orally to reduce anemia risk and enhance blood circulation. Side effects may encompass headaches, nausea, diarrhea, fatigue, rash, and fever.
- Pain-Relief Medications: Healthcare providers may prescribe narcotics to manage pain during crises.
Preventing Infections
- Penicillin: From 2 months to 5 years of age, children with sickle cell anemia may be prescribed penicillin to ward off infections like pneumonia, which can be fatal for them. Adults with a history of pneumonia or splenectomy may continue penicillin indefinitely.
- Vaccinations: Essential for all children, vaccinations are particularly crucial for those with sickle cell anemia due to the severity of potential infections. The vaccination regimen should cover pneumonia, meningitis, hepatitis B, and annual flu shots. Vaccinations remain important for adults with the condition as well.
During Pandemics
- Extra Precautions: Individuals with sickle cell anemia should exercise additional caution, such as staying home and getting vaccinated if eligible.
Advanced Treatments
- Stem Cell Transplant: This procedure may offer a cure for some children and teenagers.
- Gene Therapy: Research is ongoing to develop gene therapies that could potentially cure sickle cell disease.
Surgical and Other Procedures for Sickle Cell Anemia
Sickle cell anemia treatment extends beyond medication to include various surgical and procedural interventions aimed at managing the disease and its complications.
Blood Transfusions
- Purpose: To treat and prevent complications like stroke.
- Process: Donated blood is processed to isolate red blood cells, which are then transfused into the patient.
- Benefits: Increases the proportion of healthy red blood cells, mitigating symptoms and complications.
- Risks: Potential immune reactions, difficulty finding compatible donors, infection, and iron overload.
Managing Iron Overload
- Concern: Regular transfusions can lead to excess iron, which may harm vital organs.
- Solution: Treatments to reduce iron levels may be necessary.
Stem Cell Transplant (Bone Marrow Transplant)
- Procedure: Replaces the patient’s affected bone marrow with healthy marrow from a compatible donor.
- Potential: Can cure sickle cell anemia.
- Candidates: Generally recommended for children with severe disease manifestations.
- Risks: High, including the risk of mortality.
Stem Cell Gene Addition Therapy
- Method: The patient’s stem cells are modified to produce normal hemoglobin and then returned to the body.
- Advantage: May serve as a cure when a matched donor is unavailable.
Gene Editing Therapy
- Approval: FDA-sanctioned for individuals aged 12 and above.
- Technique: DNA in the patient’s stem cells is edited to enable the production of healthy red blood cells.
- Outcome: Successful treatment can eliminate disease symptoms.
- Status: Long-term effects are under investigation.
Ongoing Research
- Focus: Clinical trials continue to explore stem cell transplantation in adults and further gene therapies.
These procedures represent the cutting-edge of sickle cell anemia treatment, offering hope for a cure and improved quality of life. It’s important to discuss the risks and benefits with a healthcare provider to determine the best course of action. If you have any more questions or need additional information, please let me know.
Self-Care and Support for Sickle Cell Anemia
Managing sickle cell anemia involves a combination of medical treatment and self-care strategies to minimize complications and improve quality of life.
Self-Care Strategies
Nutritional Health
- Folic Acid and Vitamins: Daily supplements and a balanced diet rich in colorful fruits, vegetables, and whole grains support red blood cell production.
Hydration
- Water Intake: Aim for approximately eight glasses of water daily to prevent dehydration and reduce the risk of pain crises.
Temperature Regulation
- Avoid Extremes: Exposure to very hot or cold temperatures can trigger pain crises.
Physical Activity
- Exercise: Engage in regular, moderate exercise as advised by your healthcare professional.
Medication Use
- Pain Relievers: Use medications like ibuprofen or naproxen cautiously due to potential kidney effects.
Smoking Cessation
- Avoid Tobacco: Smoking can exacerbate the frequency of pain crises.
Coping Mechanisms and Support
Communication
- Talk Therapy: Consult a mental health expert to navigate the stress of chronic illness.
Community Support
- Support Groups: Join groups to share experiences and gain support from others facing similar challenges.
Pain Management
- Alternative Therapies: Consider heating pads, baths, massages, or physical therapy to manage pain.
Education
- Learn About the Condition: Knowledge empowers you to make informed care decisions. Utilize resources recommended by your healthcare team.
Implementing these self-care measures and seeking support can significantly aid in coping with sickle cell anemia. Remember, your healthcare team is there to guide you through this journey, so don’t hesitate to reach out to them for advice and assistance.
Preparing for Your Sickle Cell Anemia Appointment
When preparing for an appointment regarding sickle cell anemia, it’s important to be well-prepared to ensure that you and your healthcare provider can make the most of your time together. Here’s a structured approach to help you get ready:
Before the Appointment
Personal Checklist
- Symptom Record: List all symptoms, their onset, and whether they seem related or unrelated to sickle cell anemia.
- Family Medical History: Note any family members with sickle cell anemia or the trait.
- Questions for the Team: Prepare a list of questions you have for your healthcare team.
Support System
- Companion: If possible, bring someone with you to help remember the discussion.
Questions to Ask Your Healthcare Professional
- Symptom Causes: What could be causing my symptoms?
- Alternative Causes: Could there be other reasons for these symptoms?
- Diagnostic Tests: What tests will I need?
- Treatment Options: What treatments are available, and which do you recommend?
- Side Effects: What are the common side effects of these treatments?
- Treatment Efficacy: How effective is the treatment likely to be?
- Lifestyle Adjustments: Are there any dietary or activity restrictions I should follow?
- Educational Resources: Can you provide any brochures, printed materials, or website recommendations for more information?
During the Appointment
Expectations from Your Doctor
Your healthcare professional will likely inquire about:
- Symptom Timeline: When did you first notice your symptoms?
- Symptom Frequency: Are your symptoms continuous or intermittent?
- Relief Factors: Is there anything that seems to alleviate your symptoms?
- Aggravating Factors: Is there anything that appears to exacerbate your symptoms?
Being prepared with this information can facilitate a more productive appointment, helping your healthcare provider to better understand your condition and tailor the treatment to your needs. Remember, no question is too small, and it’s important to address all your concerns during the appointment.
| Category | Subcategory | Details |
|---|---|---|
| Overview | Definition | Inherited disorder affecting red blood cell shape. |
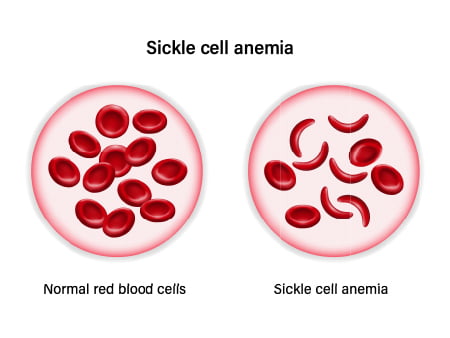
| Red Blood Cells | Normally round; sickle cell anemia causes crescent shapes. | |
| Treatment | Aimed at relieving pain and preventing complications. | |
| Symptoms | Anemia | Short lifespan of sickle cells leads to a shortage of red blood cells. |
| Pain Crises | Caused by blockage of blood flow due to sickle-shaped cells. | |
| Complications | Include swelling, infections, delayed growth, and vision problems. | |
| When to See a Doctor | Urgency | Immediate care needed for fever, stroke symptoms, and infections. |
| Causes | Genetics | Caused by a gene mutation affecting hemoglobin. |
| Inheritance | Requires both parents to carry the gene. | |
| Risk Factors | Ethnicity | Common in African, Mediterranean, and Middle Eastern descent. |
| Complications | Variety | Stroke, acute chest syndrome, organ damage, etc. |
| Preventive Measures | Genetic counseling recommended for carriers. | |
| Diagnosis | Testing | Blood tests for hemoglobin form; routine in newborn screening. |
| Stroke Risk | Ultrasound for children; regular transfusions can decrease risk. | |
| Treatment | Medications | Hydroxyurea, L-glutamine, pain-relievers, etc. |
| Infection Prevention | Penicillin and vaccinations. | |
| Advanced Treatments | Stem cell transplant, gene therapy. | |
| Procedures | Blood Transfusions | Used to treat and prevent complications. |
| Stem Cell Transplant | Can cure; high risk. | |
| Gene Therapies | Gene addition and editing therapies as potential cures. | |
| Self-Care | Nutrition | Folic acid supplements and a healthy diet. |
| Hydration | Adequate water intake. | |
| Temperature | Avoid extremes. | |
| Exercise | Regular, moderate activity. | |
| Coping and Support | Communication | Talk therapy and support groups. |
| Education | Learn about the disease for informed decisions. | |
| Appointment Prep | Checklist | Symptoms, family history, questions. |
| Expectations | Questions from healthcare professionals. |